Calibration & Validation

Clean Room /HVAC validation
Thermal validation
Compressed Air Validation
What is validation?
Clean Room / HVAC Validation
We provide an extensive range of services pertaining to clean room validation by our panel of expert professionals available with us.
SK Scientific Solutions performs the HVAC validations as per ISO 14644-1-2015, EU GMP/EC GMP, guidelines for all room classifications.
Cleanroom Certification
Validate cleanrooms as per required class of cleanliness.
The level of cleanliness chosen is driven by user requirements. Cleanroom classes are defined in ISO14644-1:-2015
Monitor And Control
A constant monitoring program is required after certification. Requirements for compliance are found in ISO 14644-2.
Statistical analysis for cleanroom parameters is encouraged as a tool for monitoring the cleanroom after certification to ensure compliance. The tool of choice is statistical process control, SPC.
Methods for evaluation and measurements for Certification are specified in ISO14644-3. It calls out for the following ten tests:
Once certified to a particular class the cleanroom factors are monitored to ensure that parameters have not drifted, or changed, and that the environment is under control.
SK Scientific Solutions carries out following tests to validate clean rooms/clean zones.
Installed Filter System Leakage Test (using PAO)
We conduct complete HEPA/ULPA filter integrity testing services. These are offered for both the Pharmaceutical and Microelectronics industries. All filter integrity tests performed by us are executed in accordance with IES-RP-CC-001-86 & ISO 14644. We are equipped to perform HEPA Filter Integrity Test with Poly Alpha Olefin (PAO) depending on client-facility’s specific requirements.
The tests assure that client’s filters are in conformance with various standards and/or governing agency requirements. Testing and evaluating filters minimum once annually and potentiality twice annually is required for optimum performance. Proper documentation and certification is provided by us and this also helps predict potential performance issues and increases filter life.
Air flow Test & ACPH Calculations
We conduct Air Velocity Measurement / tests to determine the average filter face velocity and uniformity, and the average room airflow velocity and uniformity within a clean room. The average airflow velocity is calculated by dividing the total of the airflow grid velocities by the number of readings taken. Air Exchange Rate tests determine the total air volume get in to the room within a clean room.
The use of TSI Accubalance Air Capture Hoods assures accurate test results. The hood measures air volume flowing through registers, diffusers and grills. These hoods simply hold the accubalance up to a diffuser or grill and read direct supply or exhaust airflow on the large digital display.
Non-Viable Particle Count Test
We are SK Scientific Solutions a reputed name in offering clean room validation services. Our Particle Count Test provides complete airborne particle count cleanliness classification.
The test is performed to determine the actual particle count level within the facility at the time of the test. The test identifies particle count on basis of At-Rest, In-Operational as per ISO 14644-1-2015 , EU GMP The particle size(s) of interest, the room occupancy state and the room classification shall be known prior to the beginning of the tests and shall be as specified in the URS documents.
Recovery Test
We execute recovery tests for clients across Industries. These tests demonstrate the ability of the clean room to remove particulate by purging the area with filtered air.
It also testifies if the room can change from a “dirty” to “clean” state within the specified time. The test is conducted by experienced technicians from team. Our technicians have enriching experience and provide clients with high quality service.
The ultimate goal of our company is to assure complete satisfaction of clients through effective execution of services and by providing best array of clean room equipment. We ensure that client’s clean room facility is performing properly and accurately.
Air Flow Pattern Test (Using Water Fogger)
We offer Airflow Visualization Test as a part of the validation process. Visualization is carried out by using water fogger and taking Video Graph. The purpose of the airflow visualization test is to show the actual airflow pattern throughout the unidirectional clean room. The test can also be used to demonstrate the effects on airflow caused by equipment. It is best to perform this test after all airflow velocity and uniformity tests and room pressurization tests have been performed. The test determining the airflow patterns within a room using ISO 14644-1-2015 guides. This visual monitoring service is important in:
Clean Room laminar flow tests:
Temperature & RH Measurement
We conduct validation tests that include Temperature and Humidity Measurements / Tests. Two levels of temperature and humidity tests are used by us depending on the requirement. In the first level, general temperature and humidity uniformity are tested.
The general level test is used to ensure that the clean room’s HVAC system maintains the specified levels of temperature and humidity required for occupant comfort.
The second level or the comprehensive level test identified that the clean room HVAC systems needs to maintain the specified levels of temperature and humidity required for both occupant comfort and process temperature control.
Light Intensity Measurement
The purpose of the lighting level tests is to verify that the installed light levels and uniformity meet the specified requirements.
We make use of modern testing instruments for assessment of lighting lux levels and intensity.
Sound Level Measurement
We perform noise level test that measure the sound pressure. The measurements will vary based on the occupancy state-of-the-art clean room. The purpose may vary but the procedures of testing are identical.
Thermal
SK Scientific Solutions is a leading specialist provider of validation services. We have grown as a reputable company and offer our services nationally. As an industry leader we are having more than 10 years / nearly a decade of experience using both thermocouples and thermistors.
The pharmaceutical industry is a highly regulated environment based on research, evidence, record-keeping and validation. Our experience and expertise will give you a clear, compliant and transparent validation package.
The term “Thermal validation” is the process of validating / qualifying equipment and storage facilities to prove that they will create and maintain the temperatures they are designed for.
Validation and Monitoring equipment (Wired & Wireless) are major responsibilities comes under thermal validation. Well maintained and calibrated equipment ensures that it operates at peak performance and complies with regulatory requirements.
We are specialises in the qualification of the following equipment:
Autoclave / Sterilizer Qualification
Autoclave Validation / Qualification is mandatory for all machines used for biological sterilization, in the biomedical and pharmaceutical industries within the FDA, WHO & EU controlled areas. Sterilization can be accomplished by either physical or chemical means.
This benchtop autoclave validation sterilizer. As used for sterilization of instruments and porous loads in hospitals, private clinics and dental and veterinary offices.
The principal physical means is autoclaving; other physical methods include boiling and dry heat. Chemicals used for sterilization include the gases ethylene oxide and formaldehyde and liquids such as glutaraldehyde. Of all these sterilants, autoclaving is the fastest, most reliable and hence; most commonly used within the FDA, WHO and EU zones of influence.
It must always be remembered that it is also one of the easiest processes to get wrong. This is why regulators will nearly always scrutinize and ask about validation methods used in autoclave qualification / Qualification activities.
Autoclave validation / Qualification must follow the routine validation / Qualification document string of;
VP – URS – DQ – VRA – IQ – OQ – PQ

Cold Rooms, Warehouse and Room Mapping
Cold Chain Validation suddenly became a hot topic when the results of a government investigation into Cold Chain distribution compliance were published. The facts release indicated that nearly a quarter (23 & precnt) of all drugs distributed in the USA, are of unknown efficacy. The distribution of these temperature sensitive products in vehicle containers that lacked validated methods of maintaining the correct internal temperature or even the ability to produce a validated history of what the actual conditions were; has rendered these products ‘adultered’, i.e. of unknown efficacy or safety.
Vehicle body internal temperatures; with seasonal variations, can range from a high of 50 to a low of -40 (degrees centigrade). In contrast the mandated internal temperature for distribution containers for standard drugs and medical products is 2 to 8 (degrees centigrade), with much lower temperatures specified for some drugs.
This shocking news has certainly guaranteed that Cold Chain Validation compliance will be top of everyone’s audit list, specifically when drug interactions with the end user are considered as critical.
The implications of products being damaged and out of specification may induce many companies to replace doubtful goods or even trigger the regulators into initiating product recalls. This problem is expected to create a veritable stampede as distributors scramble to ensure that all the vehicles and containers they used in this essential everyday service, are rendered Cold Chain compliant, as soon as is possible.
Depyrogenation Ovens Qualification

Dry heat sterilization (or Depyrogenation) is a process aimed at the reduction in the level of pyrogens with the use of hot air in temperature ranging from 160° C up to 400° C. The temperature used depends on the duration of the process. Gravity or mechanical heat convection can be used for this process. The former uses the natural interaction between air and different temperatures and the latter produces a specific flow of air with the help of a blower.
Depyrogenation is mainly used in the sterilization of vials for aseptic filling. The process is also useful to sterilize assembled and packaged materials, since heat conduction does not require the contact of the product with steam or water.
However, not all products can withstand the high temperatures required for this kind of process (e.g. metal goods, heat sensitive solutions, etc.).Batch and dynamic are the two main techniques for depyrogenation. Batch production is usually done in static ovens where the product is placed in metal crates and exposed to high temperatures. Dynamic involves the use of a depyrogenation tunnel, where goods are transported over a continuous band through different heating zones of the tunnel.
Lyophilizers Qualification
Freeze drying (or Lyophilization) is a technique used to remove water from a solution to leave a dry ‘cake’ as an end product. The dry product is much more stable than a solution. The reduced weight and volume is a big advantage and the removal of the water does not change the activity of the product. This technique involves freezing the product on a shelf using extremely low temperatures (typically –40 to –50°C), applying a vacuum and then slowly heating the product to ensure the ice turns into vapor. This removes the water content from product by a process called sublimation.
The critical parameters during freeze-drying are temperature (product and shelf) and pressure/Vacuum. These measurements are usually recorded with every cycle to determine when the product is dry. In addition to monitoring each cycle, freeze driers also require regular validation. This will generally involve temperature mapping of the shelves in the chamber, condenser cool down test, system evaluation test (Vacuum Pull-Down) to test Vacuum pumping system efficiency and Vacuum integrity test to show quality of seals, as well as validation of steam supplies for the SIP system.
As freeze drying is a rather expensive process most products being processed are within the pharmaceutical and biotechnology industries. Companies often use freeze-drying to increase the shelf life of products, such as vaccines and other injectables. By removing the water from the material and sealing the material in a vial, the material can be easily stored, shipped and later reconstituted to its original form for injection.
Lyophilization can also be performed in other industries like:
Food industry:
Technological industry:
Conservation and preservation industry:
Depyrogenation Tunnels
Pasteurization Tunnels
Tunnel Pasteurizers are used for heat treatment of products such as beer, soft drinks and acid preserves (fruits, acidified vegetables or pickles). etc. in closed bottles or cans. The containers are loaded at one end of the pasteurizer and passed under sprays of water as they move along the conveyor belt.
The sprays are positioned so that the containers are subjected to increasingly hot water until the pasteurization temperature is reached by the product in the bottle or can.
The containers are then gradually cooled with water until they are discharged from the end of the pasteurizer. A typical process will include most or all of the following steps:

BOD, Deep Freezers / Walk In Freezers

A freezer is a storage unit used in pharmaceutical industry to store contents such as samples and other medical material just below the freezing temperature. There are basically two styles of freestanding freezers – chest and upright models. Freezers have a long lifespan with most models having an expected lifecycle of twenty years for chest models and shorter for uprights, especially self-defrost models. There are also walk-in style freezers for bulk storage.
Steam-in-Place Qualification
Steam-in-place (SIP) cleaning uses the thermal energy of condensing steam for a controlled time to clean and disinfect the interior surfaces of bioreactors, vessels, tanks, piping, filters and associated fittings “in place,” without costly removal of the target equipment, or time-consuming disassembly of critical production lines. High-pressure steam is repeatedly introduced into the product-contacting areas of the processing system, for sufficient duration required to reduce bioburden and neutralize contaminants.
The success of the steam cleaning is validated by thermal mapping, where temperature sensors are placed throughout the system before steam-in-place cleaning to ensure that specified temperatures and exposure times are achieved. The temperature sensors or thermocouples are typically placed throughout the system via flanges and gaskets, eliminating the need to disassemble the system.
With years of experience in steam-in-place validation, Masy BioServices can help ensure the sterility of your production facility and the corresponding safety and efficacy of your products.
A pre-execution review of your production line design and evaluation of your test plan requirements will help us identify the critical areas and “problem spots” of your system, helping to ensure a successful validation of your steam-in-place sterilization.

Incubators

In microbiology an incubator is a device for controlling temperature, humidity and other conditions under which a microbiological culture is being grown.
The simplest incubators are insulated boxes with an adjustable heater, typically going up to approx. 65°C. Some incubators can go slightly higher but generally to no more than 100°C. More advanced incubators may have the ability to lower the temperature (via refrigeration) and/or the ability to control humidity or CO2 levels. This feature is specifically designed for cell growth.
Most incubators provide inner and outer glass doors for transparency. The main controller includes a timer.
Some can also be programmed to cycle through different temperatures, humidity levels, etc. Incubators can vary in size from tabletops units to units the size of small rooms. Incubators may also provide special features such as regulation of shake speed.
As for temperature, 37°C is a commonly used temperature in incubators being the normal human body temperature. Most bacteria, especially the frequently used E. Coli, grows well under such conditions.
For other applications lower temperatures are required, (for fish cells a growth temperature of 25°C is optimal).
Dry Heat Ovens
Components and products. Dry heat sterilization is often used for heat-stable oils, ointments and powders. Most often, depyrogenation of parenteral containers is performed utilizing a dry heat oven. The depyrogenation process is also utilized on certain heat-stabile components, glass containers, metal equipment, etc. to render the item and final parenteral product free of pyrogens. The equipment utilized to provide the dry heat medium must be validated to ensure that the system is able to provide sterile and/or depyrogenated components, on a reproducible basis. The validation of a dry heat sterilization and depyrogenation process involves approaches and procedures which parallel those utilized for steam sterilization. The efficiency of any heat treatment is determined by the design and source of the heat. Hot air is substantially less efficient in a thermal transfer medium as compared to steam. The validation effort must include heat distribution, heat penetration, bioburden and pyroburden determination, filter integrity and microbial/endotoxin challenges.

Furnaces

Temperature Uniformity Surveys commonly referred to as a TUS, establish objective evidence of the heat distribution characteristics throughout an oven or furnace system. It also identifies the qualified work zone within the oven or furnace system. TUS’s can also be an effective way of determining the efficiency of the furnace system and to monitor furnace performance over time.
System Accuracy Test, commonly referred to as an SAT, is another critically important and often required test. System Accuracy Tests are performed to determine the error within the temperature control system (temperature control device/lead wire/thermocouple). Regardless of the application, knowing the error that exists in your temperature control system allows for more consistent control of quality.
Stability Chamber
Stability chambers are ideal for testing storage, shelf life, packaging and stability. Stability chambers are available in temperature only or with temperature and humidity. Sizes range from reach-in to walk-in and most chambers feature a temperature range of -20° C to 60° C and an optional humidity range from 10% to 95% RH.
Stress testing is likely to be carried out on a single batch of the drug substance. It should include the effect of temperatures in 10° C increments (50° C, 60° C, etc.). Increments may be greater for accelerated testing. Stress testing for humidity typically is performed at 75% RH or greater (where appropriate).
Other tests can include effects of oxidation and photolysis. Stress testing of the drug substance can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule.
It can also validate the stability indicating power of the analytical procedures used. The nature of the stress testing will depend on the individual drug substance and the type of drug product involved.

Compressed Air Validation
Autocal has understood the importance of product risks by monitoring the air against quality standards, testing, giving accurate results and advising the risk factor to the clients. Testing and monitoring of compressed air and other process gases such nitrogen, oxygen, argon and carbon dioxide that come into direct contact with pharmaceutical products is vital to assuring the quality and safety of those products. Compressed Air validation is a critical component in the production of pharmaceutical industry and effects on the quality of the end product. It is used by operatives working in a number of industrial sectors including oil and gas, Pharmaceutical, Manufacturing, nuclear, diving and in medical applications. Compression of ambient air concentrates the normal contaminants already present in the air and can also introduce other contaminants such as Oil vapor, Condensed water and Bacteria.
Autocal validate Compressed Air in all kinds of industries as the demand for compressed air quality depends on its usage, we can also validate Compressed Air in some industries where they require extremely high quality air along with Breathing Air Validation, Instrument Air Validation and Nitrogen Gas Validation.
Compressed air is air kept under a pressure that is greater than atmospheric pressure. It serves many domestic and industrial purposes. Compressed Air is a vital energy source and is utilized in multiple operations in industries. When properly treated, Compressed air is regarded as a safe, clean utility, as compared to other energy sources. Compressed provides the energy source for pneumatic conveyers that transport liquids, powders and moisture sensitive product throughout the plant.
The Quality of Compressed air is important to ensure that product is safe. The most important parameters in specifying Compressed air quality are:
The International Standard Organisation sets quality standards for Compressed air as ISO:8573.
Dew Point

The Dew point temperature or Saturation temperature can be defined as the temperature at which Water vapour begins to condense. The quantity of any gas in a mixture can be expressed as a pressure. The major components of compressed air are nitrogen, oxygen and water vapor, so total atmospheric pressure is composed of the partial pressures of these three gases. While nitrogen and oxygen exist in stable concentrations, the concentration of water vapor is highly variable and must be measured to be determined. Gas of unknown water vapor concentration is passed over a temperature controlled surface. The surface is cooled until condensation forms. The temperature at which condensation forms is called the “Dew Point temperature.” Because there is a unique correlation between temperature and saturation vapor pressure, measuring the Dew point temperature of a gas is a direct measurement of the partial pressure of water vapor. Desiccant drying systems absorb water vapor from the air stream and can produce air with a Dew point of -40°F (–40°C) and drier if required. Dew point temperatures in compressed air range from ambient down to -112°F (–8 °C), sometimes lower in special cases.
Oil Content
A Compressed Air Oil content test is a measure of the Oil Content of Compressed air systems. The test can accurately determine the amount of Oil flowing in a Compressed Air stream and assess whether the system complies with relevant requirements. Many Compressed air systems are supplied using lubricated compressors. These Compressors often blow small particles of Oil throughout the system, which can cause a range of problems for components when the Oil emulsifies with water in the system. Oil free air is generally required for an Compressed Air System and an Oil content of less than 0.01 mg/m3 is generally specified.
Particulate Matter

Compressed air is used in a broad range of applications in the industries and used in the mixing of ingredients, cutting, sparging, drying of product, transporting/propelling product through processing systems and packaging of final product. In many of these applications, compressed air is in direct contact or indirect contact with product and the impurities in the compressed air may contaminate the product which can result in change of color and taste, reduced shelf life, in addition to exposure to bacteria and other microorganisms can result in product recalls. Compressed air, which is generated on site by pulling in ambient air and compressing it, contains water vapor, particulate matter (atmospheric air typically contains 140-150 million dirt particles/m3). More importantly, the filtration systems that are employed are designed to protect process equipment from large slugs of water, oil, rust and pipe scale with a nominal rating of 25 to 40 micron and are not capable of removing submicron contaminates. Compressed Air is generally filtered to remove Particulate Matter to a level less than 0.02 mg/m3 (size 100% <0.01 micron).
Moisture
All atmospheric air contains some water vapor which will begin to condense into liquid water in the compressed air or gas system when the air or gas cools past the saturation point, i.e., the point where it can hold no more water vapor. The condensed moisture must be removed by a separator and trap. Moisture in compressed air used in a manufacturing plant causes problems in the operation of pneumatic air systems, solenoid valves and air motors. This moisture causes rust and increased wear of moving parts as it washes away lubrication. Moisture adversely affects the color, adherence, and finish of paint applied by compressed air. Corrosion of air or gas operated instruments from moisture can give false readings, interrupting or shutting down plant processes. The malfunctioning of these controls due to rust, scale, and clogged orifices can result in damage to product or in costly shutdowns. Additionally, the freezing of moisture in control lines in cold weather commonly causes faulty operation of controls. Moisture content should be less than <0.01% for Compressed Air System.

Viable Count
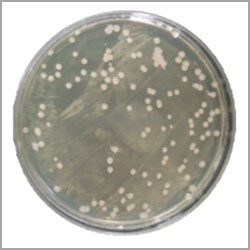
The manufacturer may not realize that the compressed air system could be harbouring microorganisms. When the air supply system is operating, it may unleash contaminants which could adversely affect the product, including operational characteristics, by compromising sterile claims, or product aesthetics. The condition or the quality of the supplied air, from a microbiological standpoint, may not be obvious unless microbiological testing is performed. A simple air impaction microbial assay of compressed air lines, however, will alert the manufacturer to the various types of viable microorganisms that might be present. Manufacturers of medical devices, pharmaceutical operations, or those classified as sterile fill, are constantly assessing the environmental impact on the product during the manufacturing process. This assessment usually includes the facility, the equipment, and the personnel involved in the assembly process. Sampling may include, but is not limited to, surface sampling, particulate counts, water analysis, and product testing. However, compressed air can easily be overlooked if not initially inserted into the environmental monitoring protocol or identified by an experienced environmental scientist. For Compressed Air System, Viable count should be <100CFU.